Posted: November 19, 2022
Content
Requirements
-
Illumina Array Analysis Platform Genotyping Command Line (iaap-cl)
-
The gtc2vcf bcftools plugin
-
Manifest and cluster files for the chip used to generate your IDAT files
Step-by-step procedure
Download iaap-cli
-
This can be downloaded from here
Note You need to create an account, and then you can download it freely.
-
Alternatively, you can run the code below. This was adapted from the gtc2vcf github page
Linux users
wget ftp://webdata2:webdata2@ussd-ftp.illumina.com/downloads/software/iaap/iaap-cli-linux-x64-1.1.0.tar.gz
Mac users
curl ftp://webdata2:webdata2@ussd-ftp.illumina.com/downloads/software/iaap/iaap-cli-linux-x64-1.1.0.tar.gz --output iaap-cli-linux-x64-1.1.0.tar.gz
Install iaap-cli
The following instructions have been adapted from the gtc2vcf github page. You may visit the github page for further useful information
First, setup some relevant paths
mkdir -p $HOME/bin && cd /tmp
Next, Extract and place iaap-cli in the relevant paths
tar xzvf iaap-cli-linux-x64-1.1.0.tar.gz -C $HOME/bin/ iaap-cli-linux-x64-1.1.0/iaap-cli --strip-components=1
Next, add $HOME/bin/ to your PATH
echo "export PATH=${PATH}:${HOME}/bin" >> ~/.bashrc
source ~/.bashrc
Otherwise, Make sure to run the command bellow each time prior to running gencall
export PATH=${PATH}:${HOME}/bin
Then, check that the installation is done properly
gencall --help
# Usage: iaap-cli gencall [arguments] [options]
#
# Arguments:
# [manifest] Location of BPM manifest file to use.
# [cluster-file] Location of EGT cluster file to use.
# [output-folder] Location to output genotype files.
#
# Options:
# -?|-h|--help Show help information
# -f|--idat-folder <idat-folder> Location of IDAT folder. Will recurse subdirectories to convert all IDAT files.
# -s|--sample-sheet <sample-sheet.csv> Location of sample sheet csv. See documentation for sample sheet format.
# -g|--output-gtc Will output GTC files for genotype info (default = false)
# -p|--output-ped Will output PED files for genotype info (default = false)
# -pt|--output-ped-tab-delimited Will output PED files delimited by tabs (default = false)
# -pcs|--output-ped-customer-strand Will output PED files using customer strand (default = false)
# -i|--gentrain-id <gentrain-id> Version of Gentrain algorithm to use (default = 3)
# -c|--gencall-cutoff <gencall-cutoff> Cutoff score for gencall algorithm (default = 0.15)
# -t|--num-threads <num-threads> Number of parallel threads to run (default = 1)
# -b|--gtc-write-buffer-size <buffer-size> Buffer size used when writing GTC files (default = 131072)
# -emi|--gender-estimate-min-loci <min-loci> Minimum number of autosomal loci for gender estimation (default = 100)
# -ema|--gender-estimate-max-loci <max-loci> Maximum number of autosomal loci for gender estimation (default = 10000)
# -emx|--gender-estimate-min-x-loci <min-x-loci> Minimum number of X loci for gender estimation (default = 20)
# -emy|--gender-estimate-min-y-loci <min-y-loci> Minimum number of Y loci for gender estimation (default = 20)
# -ec|--gender-estimate-call-rate-threshold <call-rate-threshold> Threshold for autosomal call rate for gender estimation (default = 0.97)
# -eyt|--gender-estimate-y-threshold <y-threshold> Threshold for Y intensity for gender estimation (default = 0.3)
# -ext|--gender-estimate-x-threshold <x-threshold> Threshold for X intensity for gender estimation (default = 0.9)
# -eht|--gender-estimate-x-het-rate-threshold <het-rate-threshold> Threshold for X Het Rate for gender estimation (default = 0.1)
# -inc|--include-file <include-file> Location to Loci of Interest file.
Get a pre-compiled binary of gtc2vcf plugin for bcftools
First, create a new directory for your IDAT to VCF project in a location of your choice and migrate to the direcotry.
mkdir -p idat2vcf
cd idat2vcf
Note: I always use -p to avoid overwriting a directory if it already exists
Next, check your version of bcftools by typing bcftools
bcftools
Next, go to https://software.broadinstitute.org/software/gtc2vcf/
and download a pre-compiled version of gtc2vcf by clicking on binaries for the version that corresponds to your version
of bcftools.
It will still work if your version of bcftools is higher than the version of gtc2vcf but not the other way round.
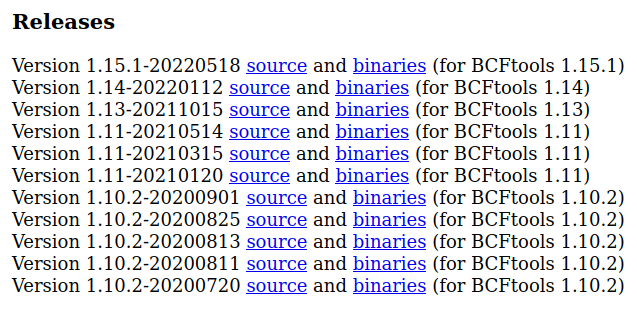
Note: Make sure your version of bcftools is greater than or equal to the version of gtc2vcf
Example
Linux users
wget https://software.broadinstitute.org/software/gtc2vcf/gtc2vcf_1.15.1-20220518.zip
Mac users
curl https://software.broadinstitute.org/software/gtc2vcf/gtc2vcf_1.15.1-20220518.zip --output gtc2vcf_1.15.1-20220518.zip
Notice that this is version 1.15.1 for bcftools version 1.15.1 or higher
Next, unzip the newly downloaded file
unzip gtc2vcf_1.15.1-20220518.zip
You should see three files in the current directory
ls
# gtc2vcf.so
# affy2vcf.so
# gtc2vcf_plot.R
We will use gtc2vcf.so to process our Illumina IDAT files
Next, test that the version of gtc2vcf downloaded works with your installation of bcftools
bcftools plugin ./gtc2vcf.so -help
# WARNING: bcftools version mismatch .. bcftools at 1.16, the plugin "./gtc2vcf.so" at 1.15.1
#
# About: convert Illumina GTC files containing intensity data into VCF. (version 2022-05-18 https://github.com/freeseek/gtc2vcf)
# Usage: bcftools +gtc2vcf [options] [<A.gtc> ...]
#
# Plugin options:
# -l, --list-tags list available FORMAT tags with description for VCF output
# -t, --tags LIST list of output FORMAT tags [GT,GQ,IGC,BAF,LRR,NORMX,NORMY,R,THETA,X,Y]
# -b, --bpm <file> BPM manifest file
# -c, --csv <file> CSV manifest file (can be gzip compressed)
# -e, --egt <file> EGT cluster file
# -f, --fasta-ref <file> reference sequence in fasta format
# --set-cache-size <int> select fasta cache size in bytes
# --gc-window-size <int> window size in bp used to compute the GC content (-1 for no estimate) [200]
# -g, --gtcs <dir|file> GTC genotype files from directory or list from file
# -i, --idat input IDAT files rather than GTC files
# --capacity <int> number of variants to read from intensity files per I/O operation [32768]
# --adjust-clusters adjust cluster centers in (Theta, R) space (requires --bpm and --egt)
# --use-gtc-sample-names use sample name in GTC files rather than GTC file name
# --do-not-check-bpm do not check whether BPM and GTC files match manifest file name
# --genome-studio <file> input a GenomeStudio final report file (in matrix format)
# --no-version do not append version and command line to the header
# -o, --output <file> write output to a file [standard output]
# -O, --output-type u|b|v|z|t[0-9] u/b: un/compressed BCF, v/z: un/compressed VCF
# t: GenomeStudio tab-delimited text output, 0-9: compression level [v]
# --threads <int> number of extra output compression threads [0]
# -x, --extra <file> write GTC metadata to a file
# -v, --verbose print verbose information
#
# Manifest options:
# --beadset-order output BeadSetID normalization order (requires --bpm and --csv)
# --fasta-flank output flank sequence in FASTA format (requires --csv)
# -s, --sam-flank <file> input flank sequence alignment in SAM/BAM format (requires --csv)
# --genome-build <assembly> genome build ID used to update the manifest file [GRCh38]
#
# Examples:
# bcftools +gtc2vcf -i 5434246082_R03C01_Grn.idat
# bcftools +gtc2vcf 5434246082_R03C01.gtc
# bcftools +gtc2vcf -b HumanOmni2.5-4v1_H.bpm -c HumanOmni2.5-4v1_H.csv
# bcftools +gtc2vcf -e HumanOmni2.5-4v1_H.egt
# bcftools +gtc2vcf -c GSA-24v3-0_A1.csv -e GSA-24v3-0_A1_ClusterFile.egt -f human_g1k_v37.fasta -o GSA-24v3-0_A1.vcf
# bcftools +gtc2vcf -c HumanOmni2.5-4v1_H.csv -f human_g1k_v37.fasta 5434246082_R03C01.gtc -o 5434246082_R03C01.vcf
# bcftools +gtc2vcf -f human_g1k_v37.fasta --genome-studio GenotypeReport.txt -o GenotypeReport.vcf
#
# Examples of manifest file options:
# bcftools +gtc2vcf -b GSA-24v3-0_A1.bpm -c GSA-24v3-0_A1.csv --beadset-order
# bcftools +gtc2vcf -c GSA-24v3-0_A1.csv --fasta-flank -o GSA-24v3-0_A1.fasta
# bwa mem -M GCA_000001405.15_GRCh38_no_alt_analysis_set.fna GSA-24v3-0_A1.fasta -o GSA-24v3-0_A1.sam
# bcftools +gtc2vcf -c GSA-24v3-0_A1.csv --sam-flank GSA-24v3-0_A1.sam -o GSA-24v3-0_A1.GRCh38.csv
Although you see that a warning is thrown that the versions of bcftools and gtc2vcf are different, the command works anyway.
Note how we call the plugin. We have used the relative path to gtc2vcf.so. It is usually convenient to use absolute paths as much as possible
To do so in our case, run pwd to get the absolute path for the current directory.
pwd
# /home/kesoh/esoh/git/esohinformatics/projects/idat2vcf
Then, run the help comman again using the absolute path
bcftools plugin /home/kesoh/esoh/git/esohinformatics/projects/idat2vcf/gtc2vcf.so -help
OR
plugin_dir="/home/kesoh/esoh/git/esohinformatics/projects/idat2vcf/"
bcftools plugin ${plugin_dir}gtc2vcf.so -help
In the second example, I have saved the abosulte path into a variable called plugin_dir, and used
the variable in the command line. This is a cleaner way of handling multiple command lines in which
the absoulte path is called multiple times.
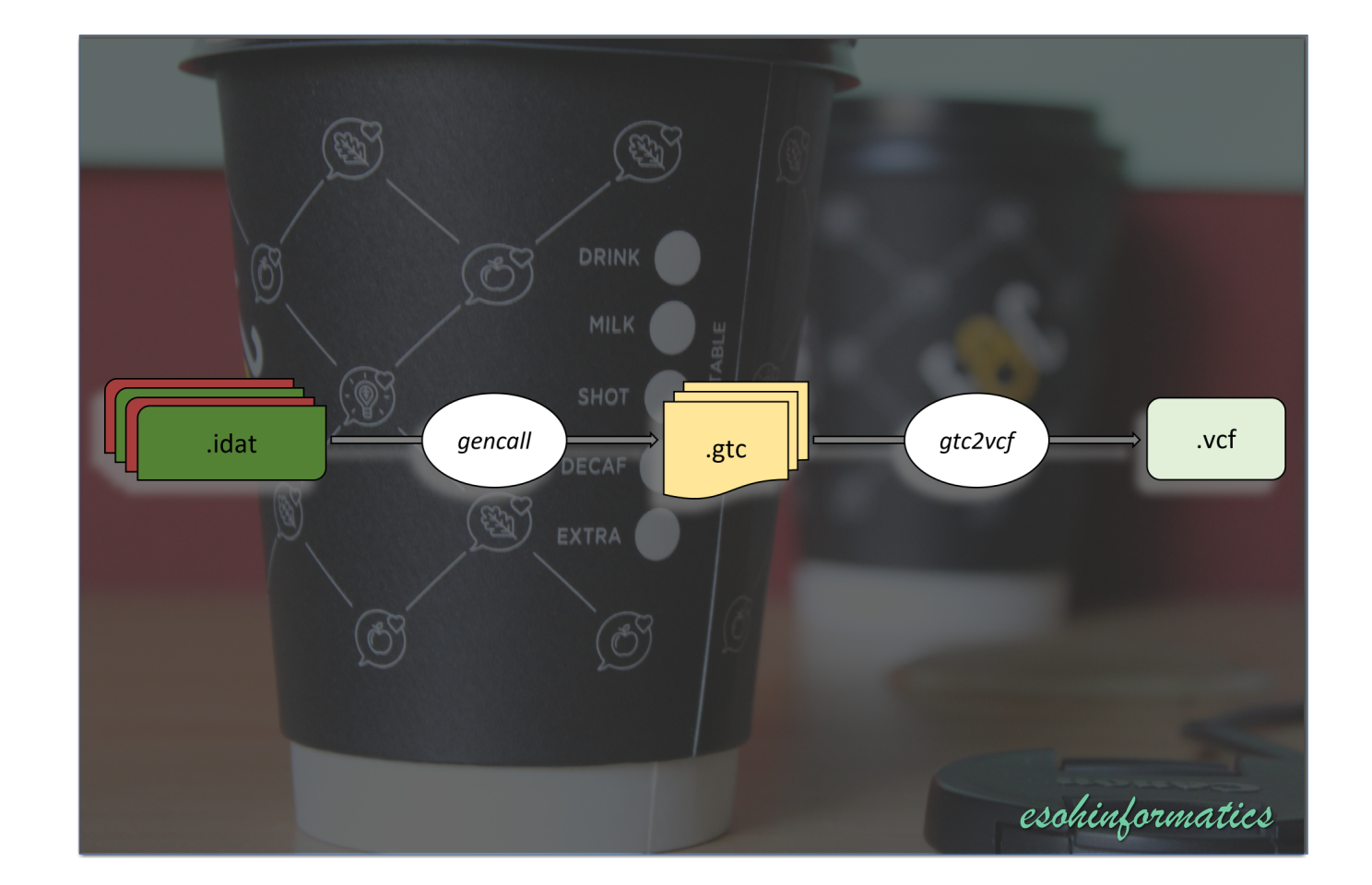
Convert Illumina IDAT files to GTC files
bpm_manifest_file=""
egt_cluster_file=""
path_to_idat_folder=""
path_to_output_folder=""
gencall \
$bpm_manifest_file \
$egt_cluster_file \
$path_to_output_folder \
--idat-folder $path_to_idat_folder \
--output-gtc \
--num-threads 10
Example: Converting IDAT files to GTC processed using the H3Africa chip
gencall \
manifest/H3Africa_2017_20021485_A3.bpm \
manifest/GenomeStudio-H3Africa-array-clusters-HapMap2-186-samples.egt \
calls \
--idat-folder intensities \
--output-gtc \
--num-threads 10 \
--gender-estimate-call-rate-threshold 0.95 \
--gender-estimate-x-het-rate-threshold 0.2
Where,
callsis the directory to which the GTC files will be saved.intensitiesis the directory containing the IDAT files. It could be a directory containing many subdirectories, each containing a number of IDAT files. IDAT files must be in pairs of red and green intensities.
Run gencall --help for more options. Choose options and adjust parameters to suit your project objectives
Convert GTC to VCF
plugin_dir="/your_gtc2vcf_plugin_directory/"
ref_dir="/your_reference_directory/"
bcftools plugin ${plugin_dir}gtc2vcf.so \
--bpm manifest/H3Africa_2017_20021485_A3.bpm \
--csv manifest/H3Africa_2017_20021485_A3.csv \
--egt clusterFile/GenomeStudio-H3Africa-array-clusters-HapMap2-186-samples.egt \
--gtcs gtcs.list.txt \
--fasta-ref ${ref_dir}human_g1k_v37.fasta.gz \
--extra summary_stats.txt | \
bcftools sort -T ./bcftools-sort.XXXXXX | \
bcftools norm \
--threads 15 \
--no-version \
-Oz \
-c x \
-f ${ref_dir}human_g1k_v37.fasta.gz | \
tee mycalls.vcf.gz | \
bcftools index \
--threads 15 \
-ft \
--output mycalls.vcf.gz.tbi
Where,
gtcs.list.txtis a list containing the gtc file names with their (absolute preferably)summary_stats.txtis a file to which some important summary statistics like gencall call rate per sample will be stored